

Karst landscapes – limestone dissolution, saturation and kinetics.
This is part of the How To…series on carbonate rocks
Mountains in traditional Chinese painting are commonly depicted as pinnacles appearing out of some ethereal mist, bound by precipitous faces; symbols of some heightened awareness, an expression of deep time. And in the fertile valleys below an alternative, ephemeral human presence, almost an afterthought.
As surreal and metaphorical as these iconic images seem, they are rooted in real-world landscapes, the karst of southern China’s Guizhou, Guangxi, Yunnan and Chongqing provinces that in 2007 were designated a UNESCO World Heritage site. Its cones, pinnacles, sinkholes, caves, bridges and Shillin (Stone Forests) developed in thick Devonian to Triassic limestone. This is one of the classic regions for tropical and subtropical karst.

Karst surface and subsurface structures are sculpted in limestone, dolostone and evaporites like gypsum and anhydrite. In all cases, the primary process is dissolution in the meteoric vadose and shallow phreatic zones. Therefore, karst is best developed in humid climates: prime examples include the tropical-subtropical landscapes of South China, temperate New Zealand (on Oligocene limestones), and the glacial – post-glacial of Ireland (the Burrens, that are underlain by Carboniferous limestone).


Karst formation provides a good opportunity to examine two competing diagenetic processes – dissolution and precipitation in terms of solution saturation, chemical kinetics, and groundwater flow.
Dissolution controlled by calcite saturation
Dissolution of limestone at the Earth’s surface is summarized in the following reaction (keeping in mind that all the carbonate equilibria are involved depending on pH, temperature, and concentrations or activities):
CaCO3 + H2O + CO2(aqueous) → Ca2+ + 2HCO3– (1)
An important determinant for calcite dissolution is the degree of saturation. Dissolution will take place in solutions that are undersaturated, precipitation in solutions that are over- or supersaturated with respect to calcite. For calcite, the degree of saturation in a solution is calculated by comparing its ion-activity product with the solubility product (i.e. the ion product if that solution were at equilibrium under the same conditions of temperature and pressure). The ratio between these two activity products is called the saturation (Ω). Ω values less than one indicate undersaturation, values greater than one over-saturation; a value of one indicates the solution is in equilibrium with solid calcite.
Rainwater contains dissolved CO2 and carbonic acid; the average pH is 5.5 to 5.8. As it filters through soils it may pick up additional CO2 from plant decay. Dissolution of limestone in the vadose and shallow phreatic zones proceeds rapidly because the water is highly undersaturated. As residence time increases in the phreatic zone, so too do the concentrations of dissolved carbonate species; there is a concomitant decrease in undersaturation and as a consequence, a decrease in the rate of limestone dissolution. At some point in this process, the saturation approaches one and dissolution ceases.
Dissolution controlled by kinetics
At this stage in our deliberations we should remind ourselves that the discussion of limestone solubility and groundwater saturation is based on thermodynamic parameters such as ion activity and energy transfer, that together determine whether a chemical reaction will proceed. What thermodynamics doesn’t do is describe the paths which these reactions take. This is the role of Chemical Kinetics. What does this mean?
Kinetics deals primarily with two parameters: the rate at which reactions take place, and the path that chemical species take to form a reaction. Reactions in aqueous solutions involve collisions between at least two ion species. They may combine directly such as:
A + B (reactant ions) → C (product)
or via a smallish number of intermediate steps until the final product is formed (these intermediate steps are called elementary reactions). Each reaction step requires that the ions have a certain amount of energy before it can proceed – this is the activation barrier.
A → A* (fast)
B → B* (slow)
A* + B* → C where A* and B* are short-lived intermediate species.
The overall rate of a reaction is determined by the slowest intermediate reaction – the one that finds it most difficult to reach its activation energy (in this case the reaction involving B).
Knowing something about the kinetics of calcite dissolution and precipitation can help us decipher which processes are important in diagenesis, whether it is cementation on the seafloor or the formation of karst.
We can now look at the picture of limestone dissolution in a different context, summarized in the diagram below. Dissolution is rapid at high degrees of undersaturation because:
- There are very few ion species competing for space on active crystal faces, and
- Dissolved mass is moved rapidly away. From a chemical kinetic perspective, the reaction is controlled by the rate at which the dissolved mass is removed from calcite crystal surfaces and transferred to some other site (transport-controlled reactions); in groundwater systems this depends on groundwater flow rates. Thus, the reaction is probably a relatively simple A + B → C type.
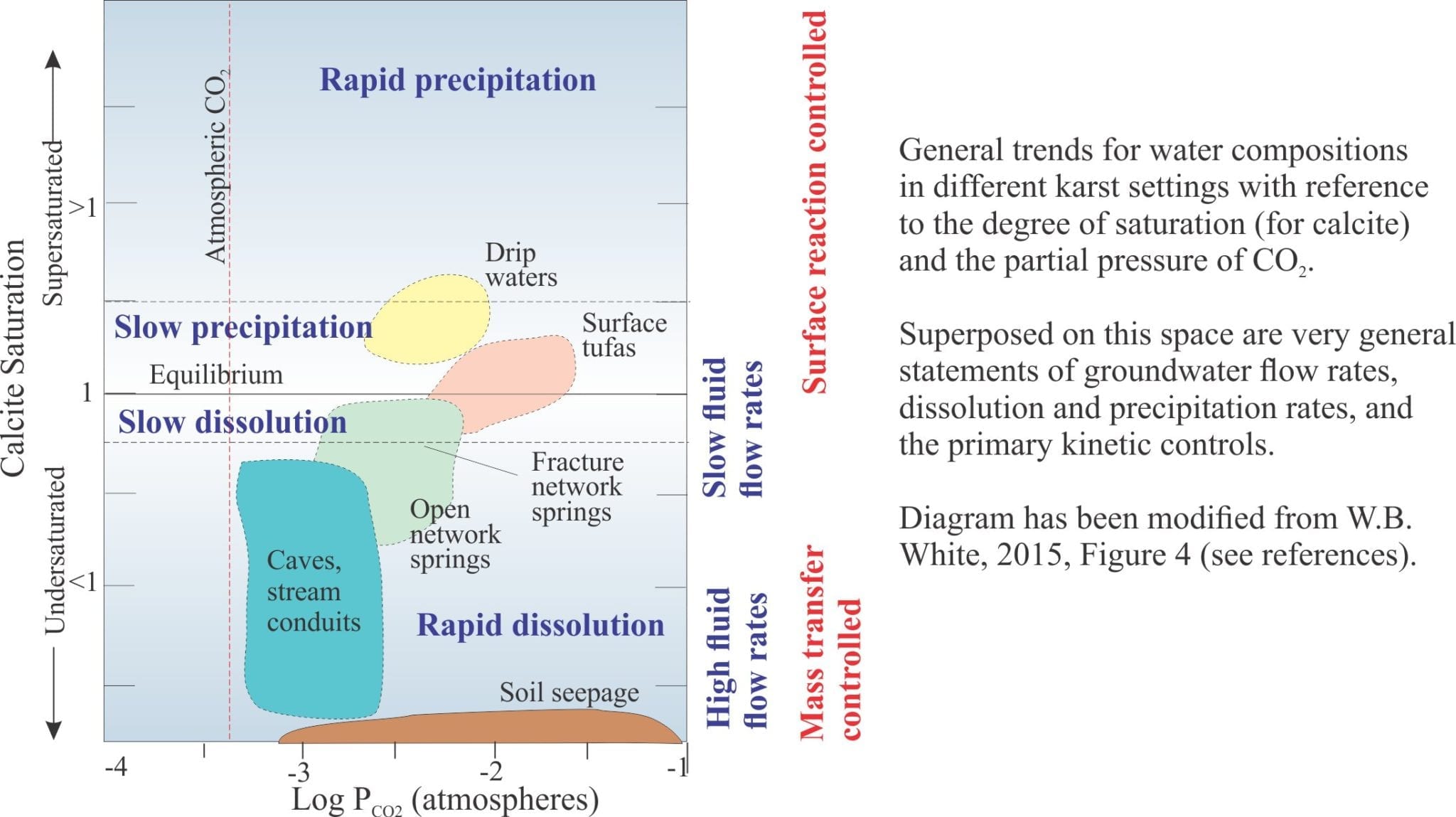
As the concentration of dissolved species increases (i.e. greater degrees of saturation) the number of molecular collisions also increases at calcite crystal surfaces. Thus, at low levels of undersaturation the rate of dissolution is controlled more by what is happening at the crystal surface – it is a surface controlled reaction and sensitive to factors such as adsorption of ion species on the surface, and dehydration of adsorbed species. For example, Ca2+ and CO32- in solution are surrounded by water molecules and for them to combine at the crystal surface, they need to shed this water (dehydrate). Under these conditions, slow intermediate reactions will determine the overall rate of dissolution.
Precipitation

At some point in their seepage journey karst fluids are capable of precipitating calcite, commonly as drip cements in caves as water filters through the vadose zone (providing us with the spectacle of stalactites and stalagmites), and in tufas where groundwaters emerge as springs. For this to happen the solutions must be over- or supersaturated with respect to calcite. However, we also know that as saturation levels approach one there is no further dissolution and therefore no mechanism to increase dissolved carbonate species to levels of oversaturation. Some other process must intervene here to produce supersaturated conditions.
It is generally understood that the partial pressure of CO2 in water passing through the vadose zone is greater than that in cave atmospheres. As water enters a cave, the various carbonate equilibria will accommodate this change in pCO2 by degassing CO2, reducing the concentration of H2CO3, and pushing reactions (1) and (2) to the left.
CO2(gas) ← CO2 (aqueous) (2)
CaCO3 + H2O + CO2(aqueous) ← Ca2+ + 2HCO3– (1)
From a kinetic perspective, calcite precipitation under these conditions is surface controlled, aided in part by the availability of nucleation sites on the crystal surface and the delivery or removal of ion species by fluid flow.
Links to other posts in this series:
Mineralogy of carbonates; skeletal grains
Mineralogy of carbonates; non-skeletal grains
Mineralogy of carbonates; lime mud
Mineralogy of carbonates; classification
Mineralogy of carbonates; carbonate factories
Mineralogy of carbonates; basic geochemistry
Mineralogy of carbonates; cements
Mineralogy of carbonates; sea floor diagenesis
Mineralogy of carbonates; Beachrock
Mineralogy of carbonates; deep sea diagenesis
Mineralogy of carbonates; meteoric hydrogeology
References and useful texts
D. Ford and P. Williams. 2007. Karst Hydrogeology and Geomorphology. John Wiley & Sons.
J.W. Morse and F.T. Mackenzie 1990. Geochemistry of sedimentary carbonates. Developments in Sedimentology 48. Elsevier, Amsterdam, 707 p.
P.A. Domenico and F.W. Schwartz, 1997. Physical and Chemical Hydrogeology, 2nd Ed. John Wiley & Sons. 506 p. This book focuses on groundwater but has an excellent section on aqueous chemistry.
USGS Glossary of karst terminology. 1972. PDF version
W.B. White, 2015. Chemistry and karst. Acta Carsologica, v.44/3, p. 349-362. Full text available