The hydrogeology and brine evolution of Death Valley waters
This is part of the How To…series on evaporites
Death Valley – you have to wonder why anyone would want to spend time there – parched, one of the hottest places on Earth, salty, life in a precarious balance. But like all deserts, quite beautiful. Extremes like these often appear other-worldly – perhaps that’s the attraction.
Death Valley is a pull-apart basin, bound on its eastern margin by the right-lateral strike slip Death Valley and Furnace Creek faults. Deformation of the basin has continued for the last 6 million years. The faults are considered to be active, with recurrence intervals of about 1000 to 2000 years. The basin is part of the Basin and Range Province that is presently undergoing lithospheric-scale extension. Slip along the Death Valley Fault System accommodates some of the strain generated at the transcurrent Pacific Plate – North American Plate boundary (Del Pardo et al. 2012).
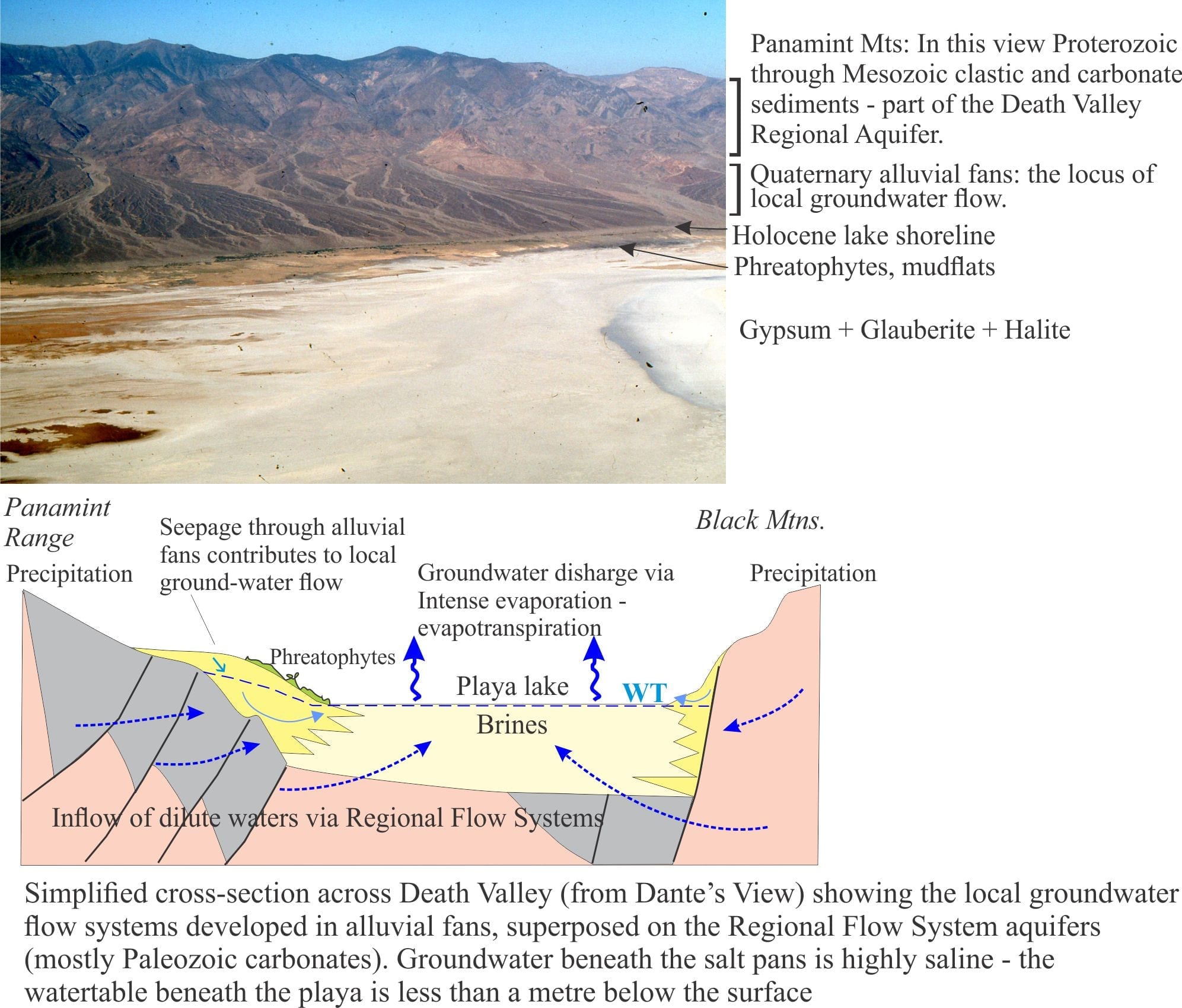
Death Valley playa lake is floored by desiccated mudflats and salt pans, the latter characterised by gypsum, halite, and glauberite. Summer maximum temperatures average 43o – 46oC but can be as high as 56oC at the aptly named Furnace Creek; winter temperatures are definitely cooler, and at high elevations are frequently sub-zero. Precipitation at the valley floor averages about 60mm/year. Precipitation increases with altitude in the adjacent ranges. Humidity is as low as 10% in the summer, and as high as 55% in winter. Thus, evaporation is intense and there is a significant deficit between water inflow and discharge.
Groundwater beneath the playa (< 1 m) is saline. However, like most saline lakes, Death Valley brines originate from dilute waters. The composition of dilute waters, whether delivered as surface runoff or groundwater, is strongly dependent on the bedrock and soil chemistry through which they flow, and in the case of groundwater the residence time for flow. Death Valley waters are interesting because three different chemical groups have been identified, depending on their association with bedrock types (Li et al. 1997; Lowenstein and Risacher, 2007):
- Na-HCO3-SO4 spring and seepage inflows that are relatively widespread in the northern and southern basins of the playa,
- Ca-Cl inflow from springs in a relatively restricted area south of Badwater, and
- Mixtures of types 1 and 2.
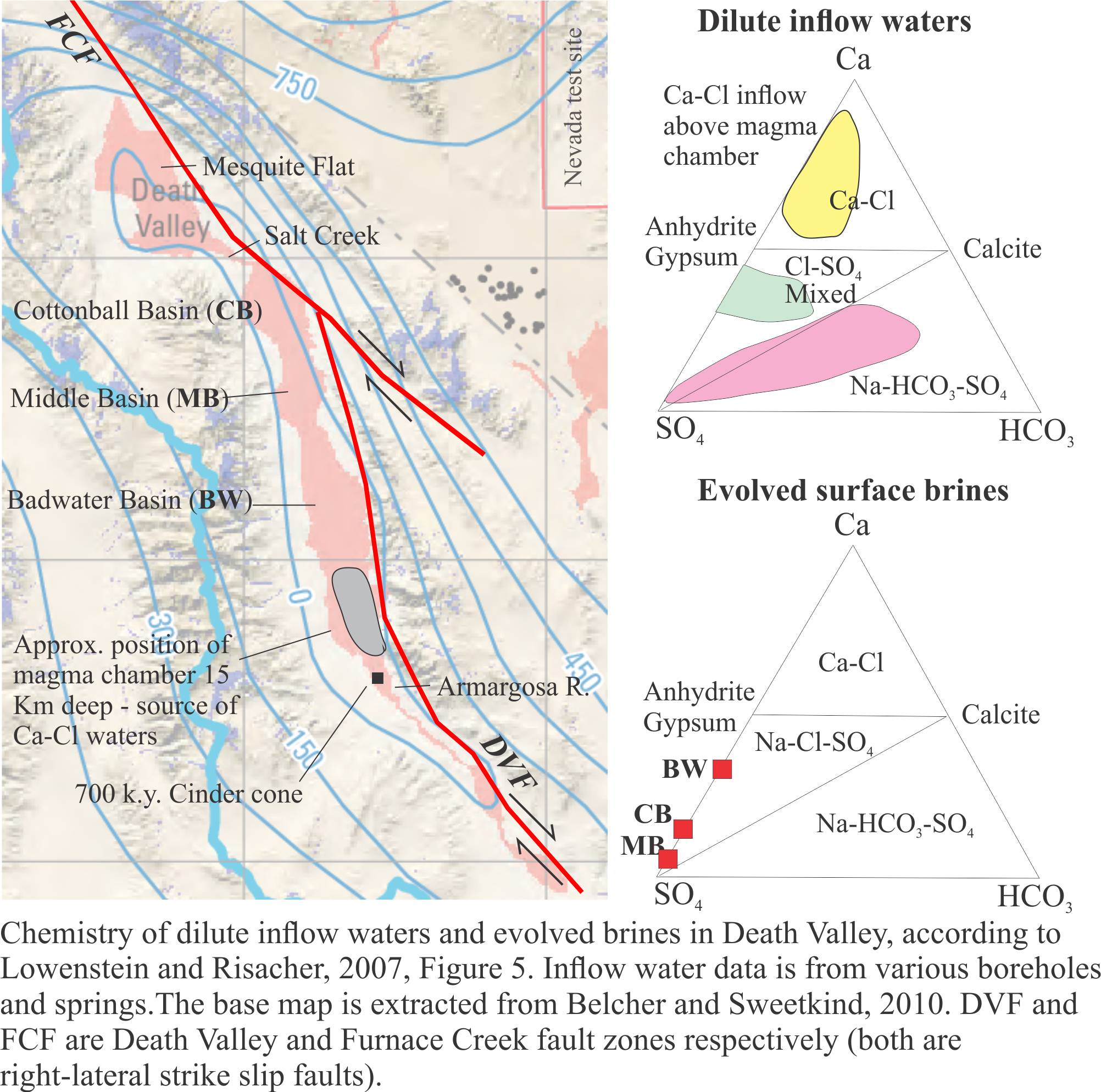
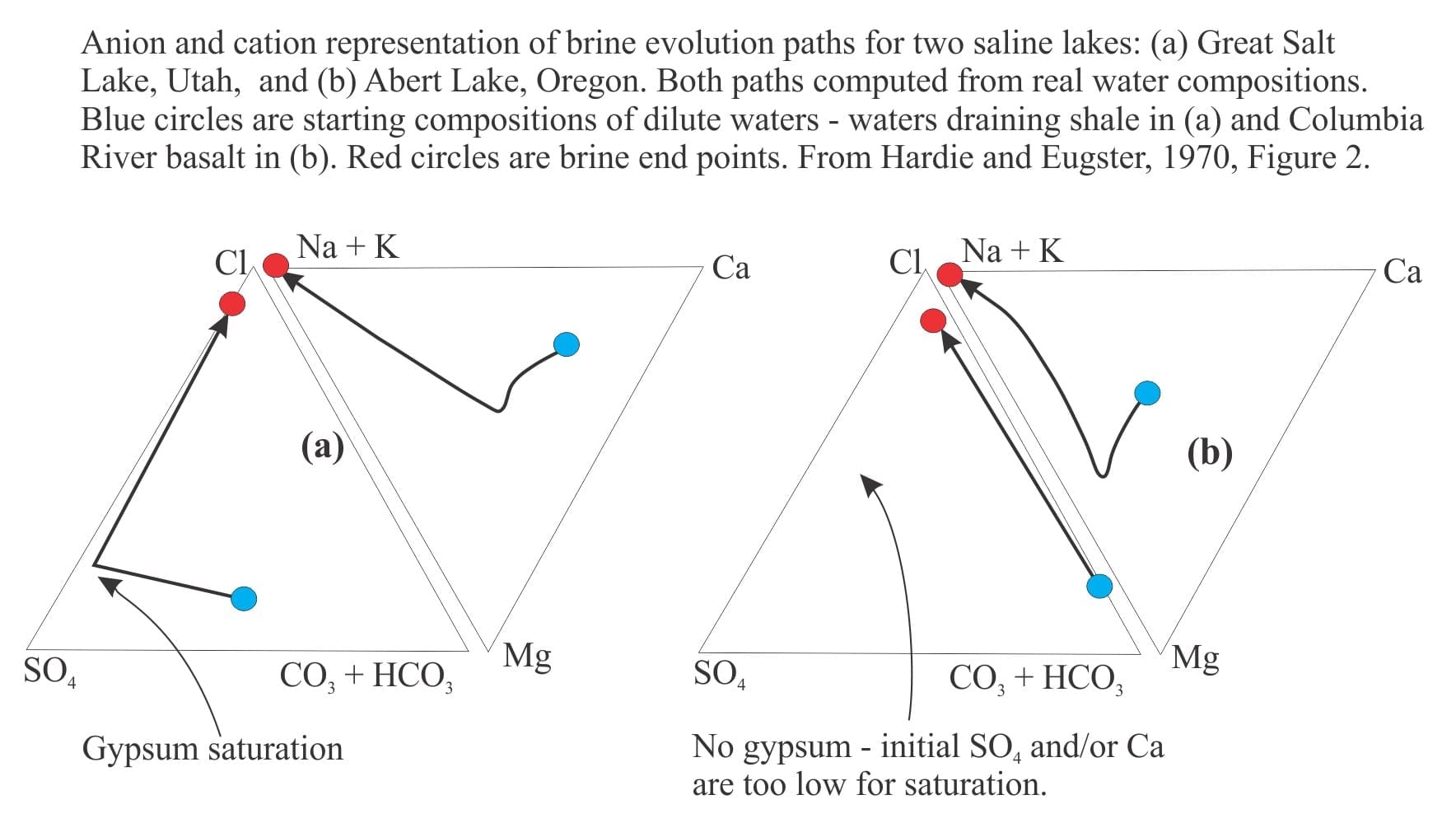
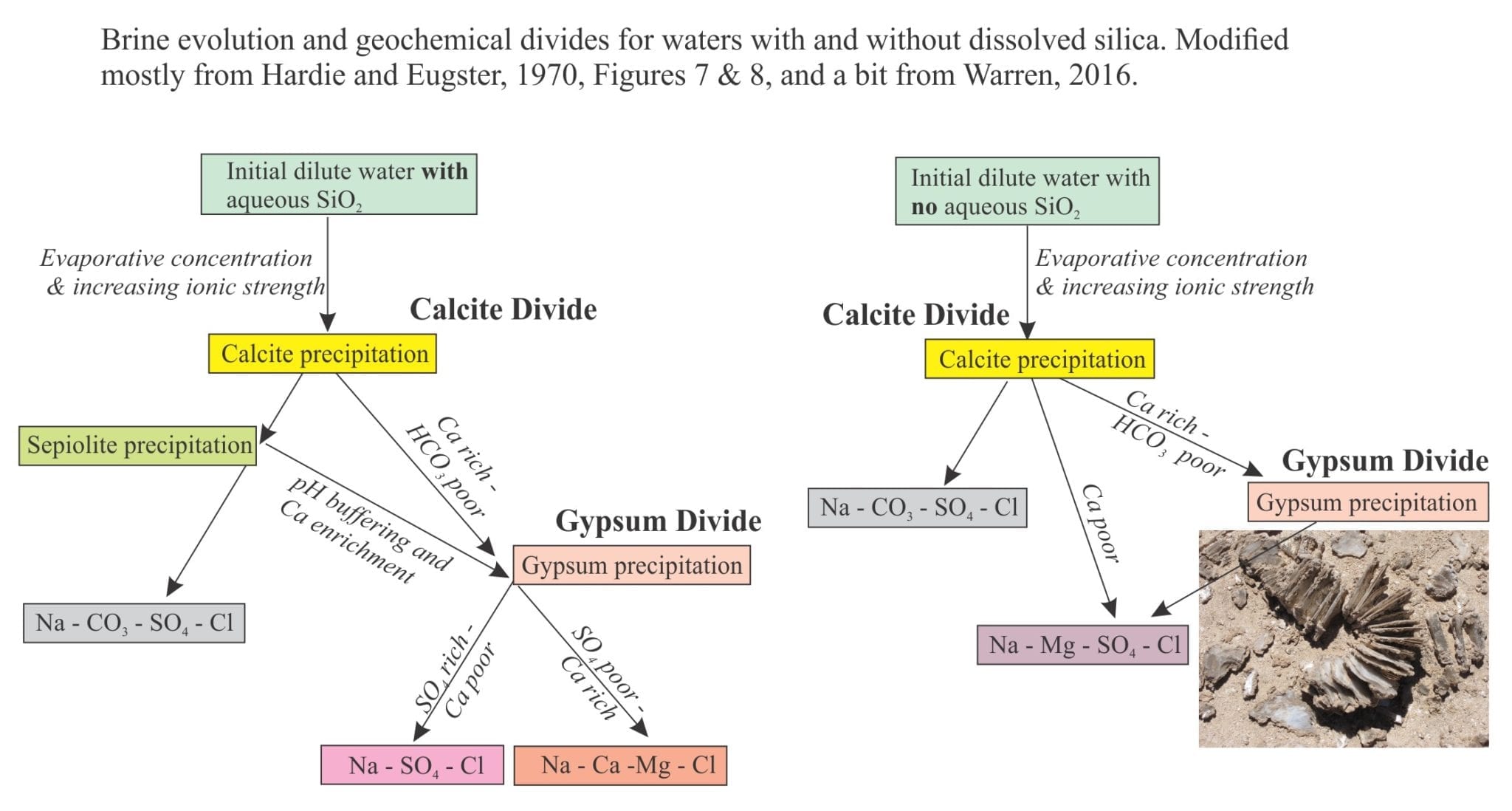
Brines produced by evaporation tend to plot in the Na-Cl-SO4 field on a Ca-SO4-HCO3 ternary diagram. According to the modelling done by Lowenstein and Risacher, brines having this composition are possible from mixing of Ca-Cl and Na-HCO3-SO4 waters. There are no pure Ca-Cl brines in Death Valley, indicating all the original Ca-Cl inflow waters are mixed.
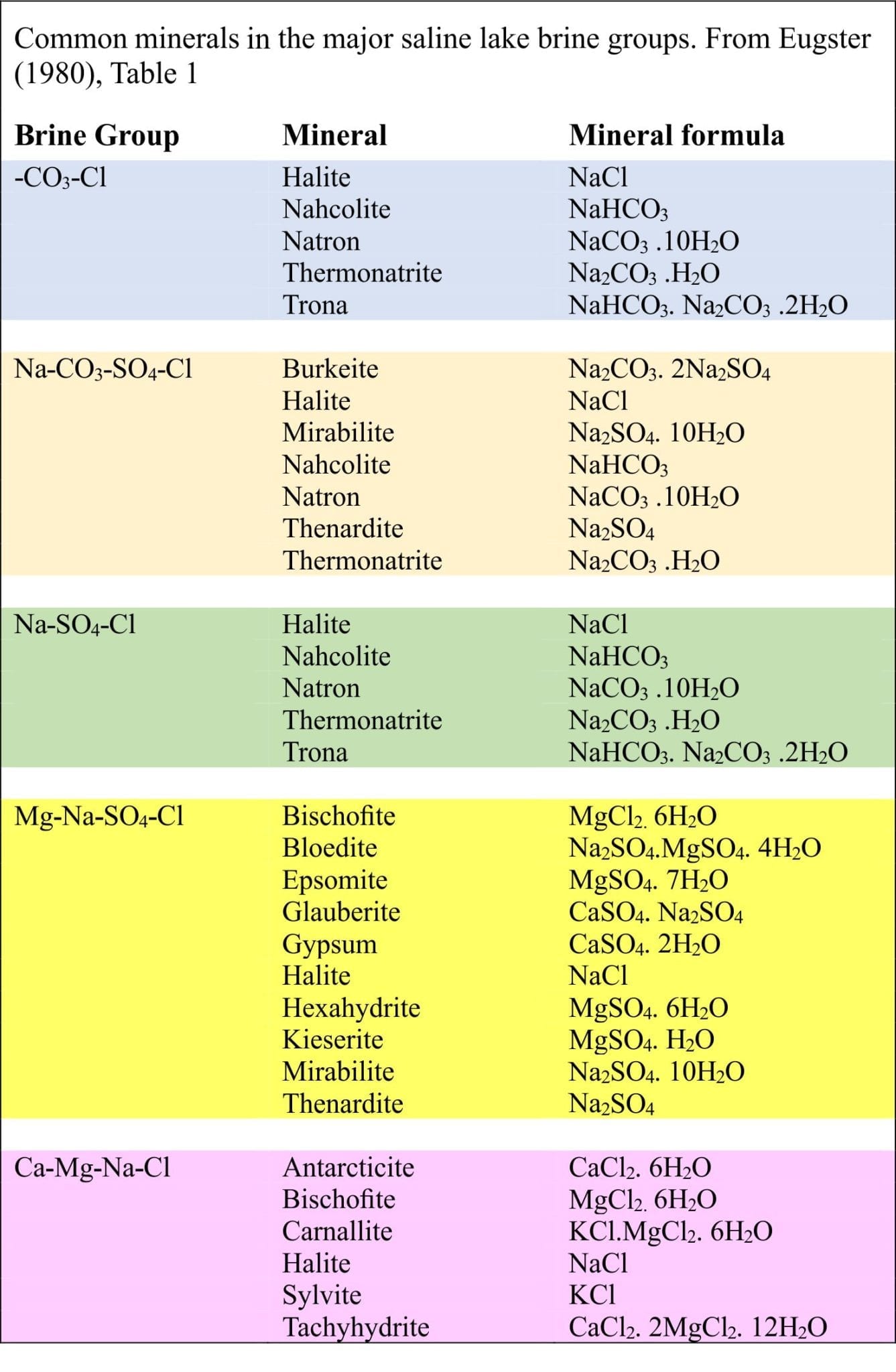
Common salts in Badwater Basin include:
- Halite (NaCl)
- Gypsum (CaSO4)
- Glauberite (CaSO4. Na2SO4)
- Borax (Na2[B4O5(OH)4].8H2O)
- Calcite (CaCO3)
Where do the dilute inflow waters that produce these brines come from?
Most of the water entering Death Valley playa lake is delivered by subsurface groundwater flow, either as diffuse flow through aquifers, or from springs. Spring flow is commonly focused along faults; perennial flows in Salt Creek, Armargosa River and Furnace Creek are spring-fed. Numerous small springs also occur at the toes of alluvial fans where they intersect the mud flats. Springs do flow at higher elevations in the adjacent ranges, but these have little influence on supply to the playa lake. Rare flash floods deliver surface runoff and sediment (commonly as debris flows or hyperconcentrated mud flows) across alluvial fans and along ephemeral streams, but this water evaporates quickly.
Phreatophytes eke out a living around springs and across parts of alluvial fans where plant roots can tap into the local watertable.
There are three main aquifers:
- Paleozoic and Mesozoic clastic and carbonate rocks that provide fracture porosity and, in the case of carbonate rocks, secondary porosity formed by dissolution of primary grains and cements. These aquifers (and aquitards) underlie the Panamint Ranges, Black Mountains, Funeral Mountains, extending to other ranges farther east.
- Pleistocene-Holocene, coarse-grained alluvial fans that drape the lower slopes of the mountains and interfinger with fine-grained playa lake deposits.
- Geothermal waters emanating from a magma body beneath Death Valley.
Investigations of Death Valley hydrogeology in the 1950s and 60s established estimates for groundwater budgets. The main components for inflowing water are groundwater, precipitation; water discharge is primarily by evaporation and evapotranspiration (i.e. via plant transpiration) – there are no natural surface flowing outlets in Death Valley. From the calculations it was apparent that there is a budget imbalance, such that the volume of water discharged was greater than that accounted for by recharge from precipitation within the Death Valley drainage basin (most aquifer recharge occurs at high elevations) and locally derived groundwater. Thus, was borne the concept of interbasin flow, where groundwater is transferred from geographically disconnected basins elsewhere in the Basin and Range Province. Differences in groundwater chemistry among the basins supports this hypothesis (see the paper by W.R. Belcher et al. 2009 for an evaluation of the basin transfer concept using the Furnace Creek springs chemistry as an example).
Interbasin groundwater transfer requires a regionally extensive aquifer system – hence the recognition of the Death Valley Regional Aquifer Flow System (an aquifer system consists of stratigraphically related aquifers and aquitards that have common permeability attributes). The aquifer system in this case includes Paleozoic and Mesozoic clastic and carbonate rocks that occur in fault-bound panels beneath the basins and enclosing ranges (shown schematically in the diagram above). The faults themselves may act as conduits or barriers to flow depending on their permeability. The regional aquifer covers an area (approximately 23,000 sq. km) about three times that of Death Valley. Recharge is mostly at high elevations in the various mountains and ranges (shown on the diagram below). Groundwater residence times in the regional aquifer are probably counted in 10s of 1000s of years, long enough to acquire the Na, Ca, K, B, SO4, Cl, CO3-HCO3, and other trace ions that characterise the Death Valley inflow waters.
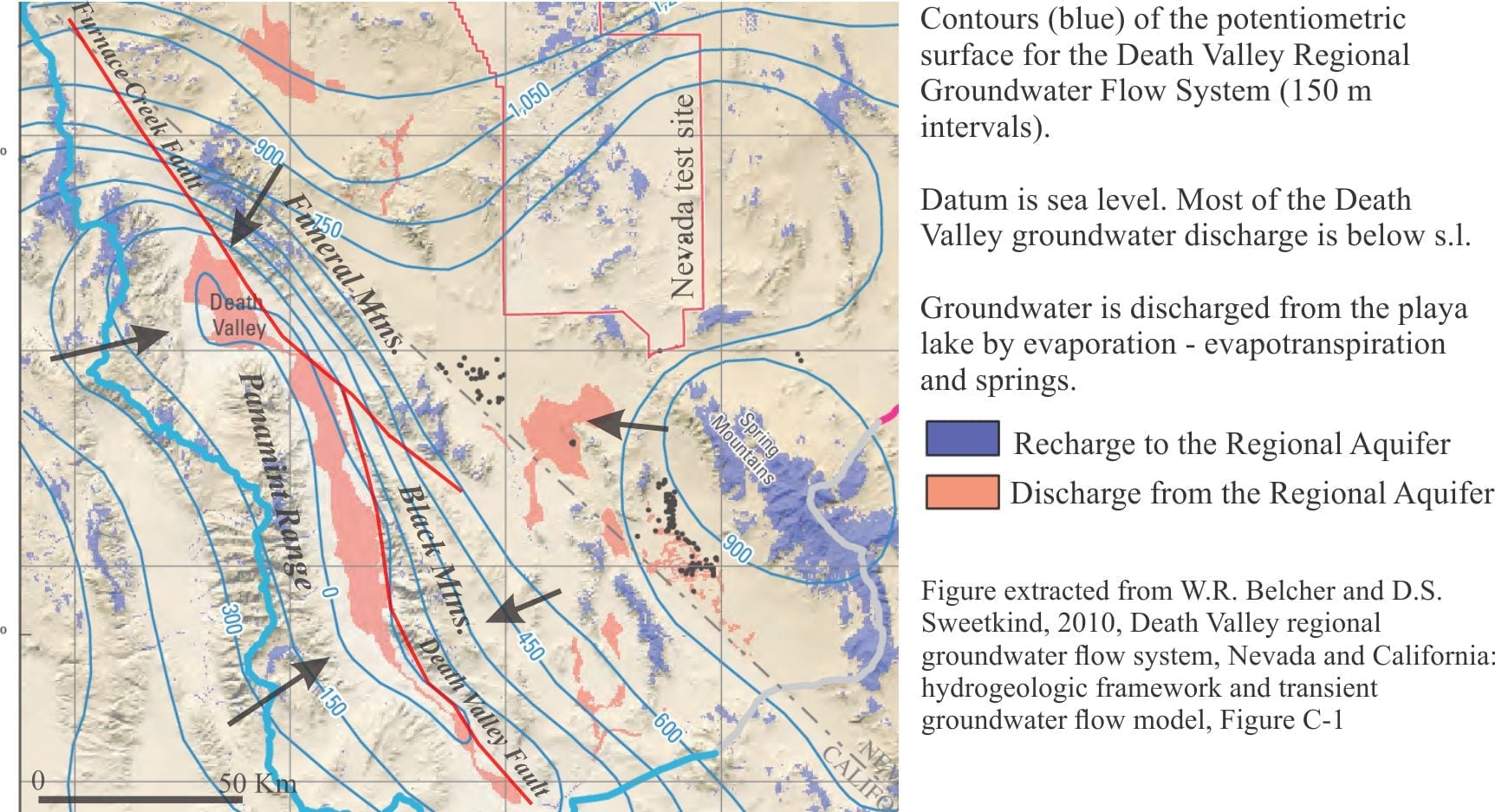
Groundwater also resides beneath the alluvial fans. Recharge to the fans is from direct precipitation and surface runoff from the adjacent ranges during winter precipitation and the occasional flash flood. The watertable is deepest at the upslope end of the fans, merging downslope with the watertable beneath the playa lake. The presence of this watertable is indicated by growth of phreatophytes. Groundwater flow through the alluvial fans comprises local, shallow flow systems that are superposed on the deeper regional flow.

The third aquifer system is that associated with a magma body, located about 15 km deep south of Badwater. The magma body has been imaged as a bright spot by reflection seismic. A 700,000 year-old basalt cinder cone nearby provides additional evidence for the magma chamber. It is hypothesized that heated Ca-Cl waters are vented to the playa lake from springs, springs that in some cases flow through faults created during magma intrusion. Ca-Cl rich waters are restricted to the area above the magma chamber where they mix with Na-HCO3-SO4 waters derived from the Regional Aquifer.
Related links
Mineralogy of evaporites: The rise of diapirs
Mineralogy of evaporites: salt tectonics
Mineralogy of evaporites: Saline lakes
Mineralogy of evaporites: Marine basins
Mineralogy of carbonates; carbonate factories
Mineralogy of carbonates; basic geochemistry
Mineralogy of carbonates; meteoric hydrogeology
Mineralogy of carbonates: sabkhas
Whiskey is for drinkin’; water is for fightin!
The Architecture of Connected Holes; A Different Way to Look at the Liquid Earth
A misspent youth serves to illustrate groundwater flow
Contrails, analogies, and visualizing groundwater flow
References
M.S. Bedinger and J.R. Harrill. 2012. Groundwater Geology and Hydrology of Death Valley National Park, California and Nevada. National Park Service, U.S. Dept. of Interior, Natural Resource Technical Report 2012/652. A comprehensive account of DV. Includes a discussion of the influence of glacial-interglacial changes in climate during the Pleistocene, the succession of lakes during pluvial dominant climates, and the alternation of evaporite – non-evaporite conditions.
W.R. Belcher, M.S. Bedinger, J.T. Back and D.S.Sweetkind, 2009. Interbasin flow in the Great Basin with special reference to the southern Funeral Mountains and the source of Furnace Creek Springs, Death Valley, California, U.S. Journal of Hydrology, v. 369, p. 30-43
W.R. Belcher and D.S.Sweetkind, 2010, Death Valley regional groundwater flow system, Nevada and California: hydrogeologic framework and transient groundwater flow model. U.S.G.S. Professional Paper 1711.
C. Del Pardo, B.R. Smith-Konter, L.F.Serpa, C. Kreemer, G, Blewitt, W.C. Hammond. 2012. Interseismic deformation and geologic evolution of the Death Valley Fault Zone. Journal of Geophysical Research, v. 117
Li, J., Lowenstein, T.K. and Blackburn, I.R. 1997. Responses of evaporite mineralogy to inflow water sources and climate during the past 100 k.y. in Death Valley, California. Geological Society of America Bulletin, 109, p. 1361-1371. The paper that identifies three chemical groups of inflow waters.
T.K. Lowenstein and F. Risacher, 2009. Closed basin brine evolution and the influence of Ca-Cl inflow waters: Death Valley and Bristol Dry Lake, California, Qaidam Basin, China, and Salar de Atacama, Chile. Aquatic Geochemistry, v. 15, p. 71-94. Focuses on the Ca-Cl waters originating from a magma chamber beneath DV. Also describes briefly the changes wrought by recurring pluvial periods during the Pleistocene.